Compliance for Medical device registration & regulation Landscape in EU
The European Union represents one of the most highly regulated and commercially attractive medical device markets in the world. Together with 27 member states, 3 EEA countries and Turkey, union market is operating under a harmonised Regulatory ecosystem defined by EU MDR (2017/745) for medical devices and EU IVDR (2017/746) for in vitro diagnostics, the region ensures uniform safety and performance standards across all product categories. To legally place a device on the EU Union market, manufacturers must obtain CE marking, which confirms compliance with the applicable regulation based on the device’s risk classification and conformity assessment route. Failure to meet these requirements can result in serious consequences, including product recall, seizure at customs, suspension of certification, or complete loss of market access.
Under the current Regulatory regime:
- CE marking is mandatory for all medical devices and IVDs before they can be commercialised.
- EUDAMED device registration is being introduced in phases, and several modules are already active for mandatory use.
- Appointment of a European Authorised Representative (EAR) is obligatory for any manufacturer located outside the EU/EEA/Turkey.
- Stricter MDR/IVDR classification rules demand robust technical documentation, clinical or performance evidence, and continuous lifecycle monitoring.
Freyr supports manufacturers throughout this journey by enabling smooth and compliant entry into the Union market. From developing a CE marking strategy and compiling technical documentation to managing Notified Body submissions and acting as your EU Authorized Representative, Freyr ensures fully aligned MDR/IVDR compliance and seamless access to all EU member states, EEA regions and Turkey.
Step-by-Step EU Compliance Process
Market access of a medical device &IVD in the EU/Union market involves several defined stages. Here’s how Freyr manages the entire process for you:
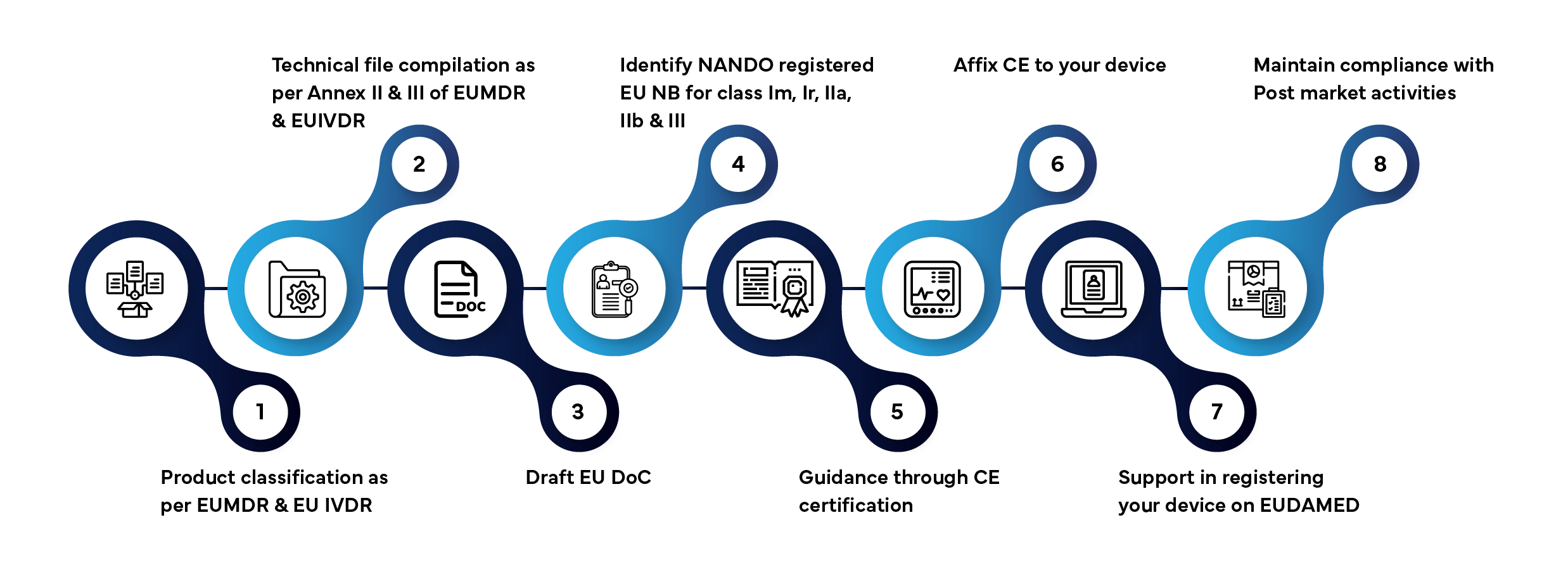
Typical processing time: 3–12 months, depending on device class and documentation readiness.
Freyr Medical Device EU Key Offerings
- Regulatory Strategy & MDR/IVDR Transition – We craft end-to-end regulatory roadmaps tailored for the EU MDR and EU IVDR, ensuring smooth migration from Directive regimes and alignment with current EU requirements.
- Technical Documentation & Conformity Assessment – Our team supports the development, review and submission of your Technical Files / Design Dossiers, assists with Notified Body engagements, and manages CE-Marking , device safety & performance test support and conformity pathways.
- Clinical/Performance Evaluation : Freyr provides expert preparation of CERs, PERs, PMCF/PMPF plans, PSURs, SVR, CPR for IVDs ,risk documentation, and biological evaluation content, ensuring technical clarity and regulatory accuracy across all device classes.
- UDI & EUDAMED Registration Support – We ensure your Unique Device Identification (UDI) system is compliant and assist with registration in the European device database EUDAMED and associated lifecycle management.
- European Authorized Representative (EAR) & Local Representation – For manufacturers outside the EU/EEA/Turkey, we act as your mandated European Authorized ReprThis aesentative (EAR) and provide local compliance support across EU member states.
- Post-Market Surveillance (PMS)– Freyr supports you to establish, implement and maintain PMS systems including Post-Market Surveillance Plan (PMSP), Post-Market Surveillance Report (PMSR), Periodic Safety Update Report (PSUR), Post-Market Clinical/Performance Follow-Up plans (PMCF/PMPF), vigilance reporting, FSCAs, FSNs, CAPAs ensuring continuous CE-mark retention and sustainable market access with product lifecycle compliance.
- CE Device Registration:Freyr manages the complete EU CE-marking registration pathway by supporting conformity assessment, compiling compliant submissions, engaging with Notified Bodies, and ensuring timely approvals across all medical device and IVD categories.
- QMS Support:We support the implementation and maintenance of ISO 13485-compliant Quality Management Systems, aligned with the EU MDR/IVDR Quality and Safety requirements and Notified Body expectations.
- Labeling Compliance:Our team ensures your labeling, IFUs, packaging, and symbols meet MDR/IVDR and EU multilingual language requirements, maintaining consistency and compliance across 27 EU member states.
EU Authorized Representative (EAR) Service Offerings:

Device Registration with EU Authorities
For non-EU manufacturers, Freyr serves as the appointed EU Authorized Representative (EAR) in accordance with MDR/IVDR requirements. As a Germany-based entity, Freyr supports device registration activities with the relevant German Competent Authority and maintains required regulatory records. For other EU Member States, Freyr provides regulatory support, as applicable, to help ensure device compliance and facilitate placement of the device on the Union market.

Documentation & Conformity Assurance
Our regulatory experts verify that your Declaration of Conformity (DoC), CE Certificates, Technical Files are complete, current, and compliant with MDR/IVDR. Freyr ensures full readiness for conformity assessment and CE marking submissions.

Responding to Competent Authority Queries
If required Freyr handles all direct communication and clarification requests from EU Competent Authorities or Notified Bodies on your behalf, ensuring timely, accurate responses and helping prevent delays to approvals or post-market regulatory reviews.

Vigilance & Incident Communication
As your EAR, Freyr acts as the primary liaison for safety-related communications. If and when required, we coordinate incident notifications, Field Safety Corrective Actions (FSCA), and vigilance reporting between the manufacturer, healthcare professionals, and authorities to ensure proper response and regulatory alignment.

Inspection & Audit Readiness
Freyr maintains all documentation, correspondence, and mandatory records for authority audits and inspections. Our team ensures that Technical Documentation, labeling and post-market records are readily available and fully compliant with MDR/IVDR expectations.
Why Partner with Freyr?
- End-to-end regulatory expertise that spans from pre-market registration to post-market vigilance, managing every stage of compliance.
- A proven track record with 2000+ device registrations completed successfully across diverse categories.
- Strong EU presence anchored in Germany (EAR), complemented by on-ground regulatory specialists in Reading and supported by Freyr’s global delivery teams.
- Tailored transition planning that provides strategic support to obtain CE smoothly and cost-effectively.
- Multi-region delivery teams with on-ground support through Freyr’s Germany-based EU operations.
- Proven track record in MDR/IVDR remediation and legacy device transition
- Transparent project governance with direct Notified Body communication